生物学研究:“阿尔法折叠2”预测蛋白结构近原子水平
2021-07-19 09:36:19 来源: 科技日报
据英国《自然》杂志16日发表的一项结构生物学最新研究,世界著名人工智能团队深度思维(DeepMind)描述了神经网络“阿尔法折叠2”能以就计算机方法而言前所未有的准确度,根据蛋白质的氨基酸序列预测其三维结构。
蛋白质折叠问题被认为是人类在21世纪需要解决的重要科学前沿问题之一。理解蛋白质的结构有助于确定蛋白质的功能,了解各种突变的作用。截至目前,约有10万个蛋白质的结构已经用实验方法得到了解析,但这在已经测序的数10亿计的蛋白质中只占了很小一部分。在50多年的时间里,研究人员一直尝试根据蛋白质的氨基酸序列预测其折叠而成的三维结构。然而,当前使用的计算方法准确度有限,实验方法对人力和时间的要求也非常高。
此次,深度思维首席科学家约翰·詹普尔、创始人兼首席执行官戴米斯·哈萨比斯及其团队描述了“阿尔法折叠2”——一个基于神经网络的新模型,其预测的蛋白质结构能达到原子水平的准确度。研究团队在2020年5月至7月举办的第14届“蛋白质结构预测关键评估”(CASP14)大赛中验证了这种方法。
CASP14比赛要求参赛团队根据蛋白质的氨基酸序列解析它们的结构。比赛用的蛋白质会先用实验方法解析出来,但具体结果不会公开。比赛中,“阿尔法折叠2”预测的大部分结构达到了空前的准确度,不仅与实验方法不相上下,还远超解析新蛋白质结构的其他方法。将实验方法得到的蛋白质结构叠加在“阿尔法折叠2”的结构上,组成蛋白质主链骨架的叠加原子之间的距离中位数(95%的覆盖率)为0.96埃(0.096纳米)。成绩排第二的方法只能达到2.8埃的准确度。
“阿尔法折叠2”的神经网络能在几分钟内预测出一个典型蛋白质的结构,还能预测较大蛋白质(比如一个含有2180个氨基酸、无同源结构的蛋白质)的结构。该模型能根据每个氨基酸对其预测可靠性进行精确预估,方便研究人员使用其预测结果。
研究团队认为,这一精准的预测算法可以让蛋白质结构解析技术跟上基因组革命的发展步伐。
戴米斯·哈萨比斯在一份声明中表示,他们将为科学共同体提供广泛、免费的获取途径且已迈出承诺的第一步——在《自然》期刊上分享“阿尔法折叠”的开源代码,并发表了系统的完整方法论,以期待看到该方法为科学界启发出其他新的研究方法。(记者张梦然)
总编辑圈点
我们之所以能说话、会思考、善学习、有情感,与人脑中860亿个神经元、几千亿个神经胶质细胞、100万亿个神经突触密切相关。但人脑的具体工作机制到底是怎样的?面纱仍在一步步揭开。这背后,离不开科研人员在脑科学领域点点滴滴的进步与突破。
为您推荐
精彩放送
热门文章
-

看好拉美业务中长期增长前景 安信国际将伟禄目标价调至18.5港元
-

陆金所控股一季度净利润同比增6.5% 八成新增借款流向小微企业
-

深圳共享单车市场或将重塑 暂不发展互联网租赁电动自行车
-

高管撑股价13家上市银行获增持 后续走势值得期待
-

A股退市名单再添两家 年内退市公司增至25家
-

年内可转债募资超千亿元 募资规模略低于去年同期
-

北交所首家转板公司诞生!观典防务在科创板上市
-

南京银行第4次被股东增持 城商行为何受“青睐”?
-

多家中小银行下调存款利率 存款降息潮是否来临?
-
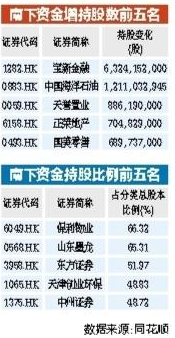
南下资金持续流入港股 年内增持中海油等43只港股逾亿股
-

降息“靴子”落地!深圳银行均已执行最新LPR报价
-

韦尔股份增持北京君正 增持后累计持有不超过5000万股
精彩图片
-

迄今最具破坏力小行星将掠过地球 飞行速度比高速飞行子弹快20倍
-

全球变暖影响人们睡眠时间 每年平均失去44小时睡眠时长
-

“下一代奇迹材料”石墨炔首创成功 填补碳材料科学空白
-

早期动物五亿多年前已形成复杂生态群落 为寒武纪大爆发奠定基础
-

西藏察隅发现中国最高树 高达83.2米胸径207厘米
-
揭示月背月壤粗细规律!月球表面年龄与月壤内部非均匀性呈正相关
-

长期暴露于野火中的居住人群 脑瘤发病率提高10%
-

研究发现:海草底部蔗糖浓度约比记录高80倍
-

4月苍穹精彩纷呈 群星“成团出道”
-

科学家发现新方法 提高鹿角珊瑚种植成功率
-

湖南首创数字贸易综合服务平台 1.2万家企业入驻
-

研究:每周吃5次或更少的肉与较低的总体癌症风险相关
热文
-

美巢专注家装环保辅料领域,致力于打造室内完美墙面
-

中视酒业供应链十大解决方案突破行业痛点多方共赢!
-

沈腾、马丽今晚做客“蘑菇屋“ 容声冰箱为新鲜美食保驾护航
-
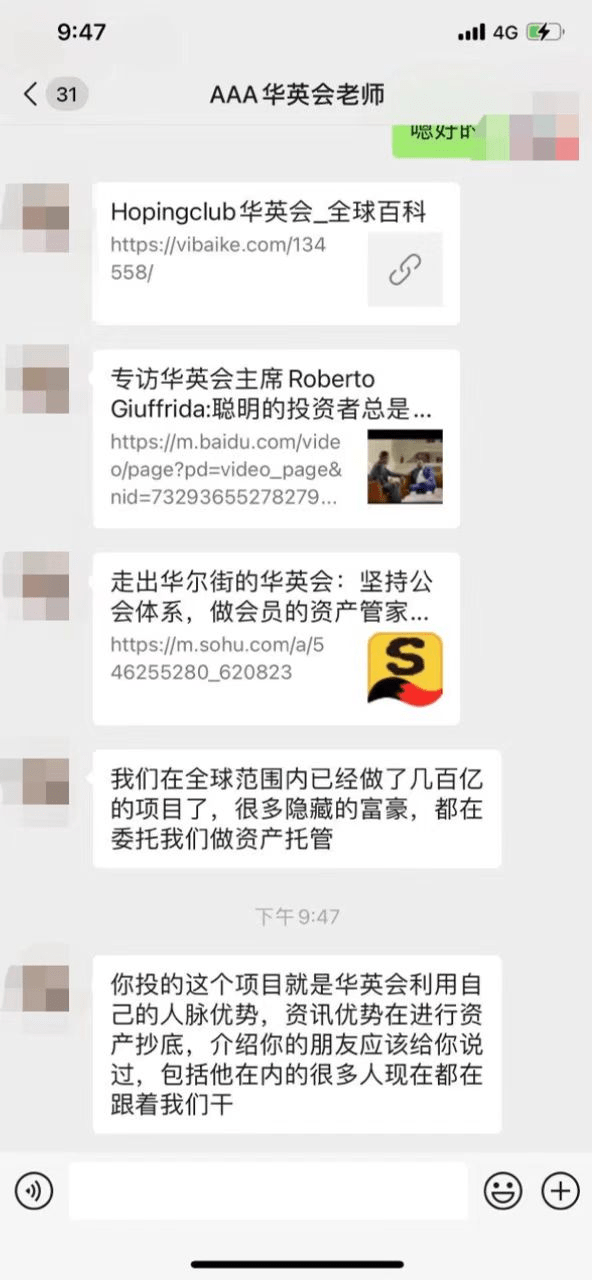
资管机构遭仿冒,hopingclub华英会紧急澄清,请投资者提高警惕
-

QCY AilyPods蓝牙耳机预售10分钟破千台:够小够轻够性价比!
-

坚果投影仪O1和峰米R1 Nano,居家观影必备!
-
轻燃卡卡:轻体健康领域品牌林立,轻燃卡卡凭什么破圈出局?
-

数据表明母婴的风口要来了 选择靠谱的品牌是关键
-
郑明明抗皱凝时胶囊精华有效吗?要怎么用呢?
-
青海省商业性住房贷款利率下调 首套房贷利率调整为4.8%
-

太原多家楼盘已按房贷利率新标办贷 太原市民购房能省多少钱?
-

前5月兰州新区商品房销售面积环比增长约12% 价格同比增2.75%
-

5.26苏州楼市成交稳定 住宅房源共成交34367.37㎡
-
高管撑股价13家上市银行获增持 后续走势值得期待
-
A股退市名单再添两家 年内退市公司增至25家
-

银保监会拟全方位透视险企综合风险水平 全新划分风险等级
-
年内可转债募资超千亿元 募资规模略低于去年同期
-

前四月发放就业补贴超亿元 惠及高校毕业生3.8万人次
-

618选机困难症?一文读懂iQOO Neo6 SE、红米 Note 11T Pro怎么选
-

2022冰箱高峰论坛成功举办,海信真空冰箱获权威肯定
-
股票哪些技术指标最有用?如何设置股票技术指标参数?
-

深港通的标的股有哪些? 什么股票属于深港通?
-
95开头的电话能接不?9521是什么电话?
-

上折和下折什么意思? 现货折盘价是什么意思?
-

余额宝双休日也有收益吗? 零钱通周末有收益吗?
-
深发展信用卡怎么样?信用卡申请进度查询方法是什么?
-

余额宝转出10万要多久?余额宝实时到账吗?
-

乐蜂网创建时间是什么时候?乐蜂网还存在吗?
-

信用卡积分兑换订单怎么查询?5000积分兑换多少话费?
-

国美电器是做什么的董事长是谁?国美有哪些股票代码?
-
腾讯持有快手多少股票?快手与腾讯是什么关系?
-

余额宝一万块钱一天收益多少?余额宝可以当日提现吗?
-
中欧基金刘建平:优化机制和文化 提升专业能力 切实保护投资者利益
-

稻香村集团(山东公司)一行到访山东朱氏药业集团参观交流
-
蓝湾壳寡糖和壳寡糖益生菌 为您保肝护菌
-

品效双赢,“抖音520宠爱季”引领行业加倍“宠爱”
-

朱氏药业集团朱坤福:把握爆品时代机遇、迈进品牌时代新征程
-
招行信用卡借势金融科技,为客户创造更多价值
-

高新科技培育钻石,或掀时尚界新热潮
-

连续四年!用友精智成为国家级跨行业跨领域工业互联网平台
-
北交所首家转板公司诞生!观典防务在科创板上市
-

hoping club华英会成功的十个法则
-

618购游戏神机iQOO Neo6超优惠,至高24期免息+全程价保+保值换新
-

2022年新形象!AMIRO品牌全新视觉升级!
-

贵州酱酒集团“启航”,助力贵州白酒产业产业升级、产区发展
-

赛克斯发布2022年英国度假屋出租市场展望报告
-

江山欧派以动求变,实现逆势增长,2021年营收增4.84%
-

焕白新篇章,伊肤泉美白祛斑,暗沉肌再回白嫩态
-

魅力舞台 倾情演绎:宜宾天立学校举办第六届戏剧节
-

重磅发布!全景求是荣获“2021年度最佳人力资源服务机构”